Table Of Content

Specifically, patients are grouped into strata according to the important factors and then randomized within each stratum. For considerations on other study designs, including adaptive, group sequential and Bayesian designs, see Pallmann et al. (2018), Bhatt and Mehta (2016), Vandemeulebroecke (2008), Lee and Chu (2012), and Berry (2006). For considerations when drafting a statistical analysis plan for clinical trials, see Gamble et al. (2017).
FDA Clinical Investigator Training Course (CITC) 2023 - 12/06/2023 - FDA.gov
FDA Clinical Investigator Training Course (CITC) 2023 - 12/06/2023.
Posted: Wed, 06 Dec 2023 08:00:00 GMT [source]
The I-SPY COVID Consortium
In the remainder of this paper, we frame our guidelines around these phases in the design process of a clinical trial. Throughout the paper, we provide references for the interested reader to find further details and explanations of concepts and terms. What are the lessons learned from this trial experience that may have important implications for future pandemics and/or clinical trials in a similar treatment space? First, there is a unique niche for phase 2 clinical trials that can rapidly evaluate repurposed or novel agents for which preliminary safety data exist, but fewer data are available in support of efficacy than would be advisable in a standard phase 3 study.
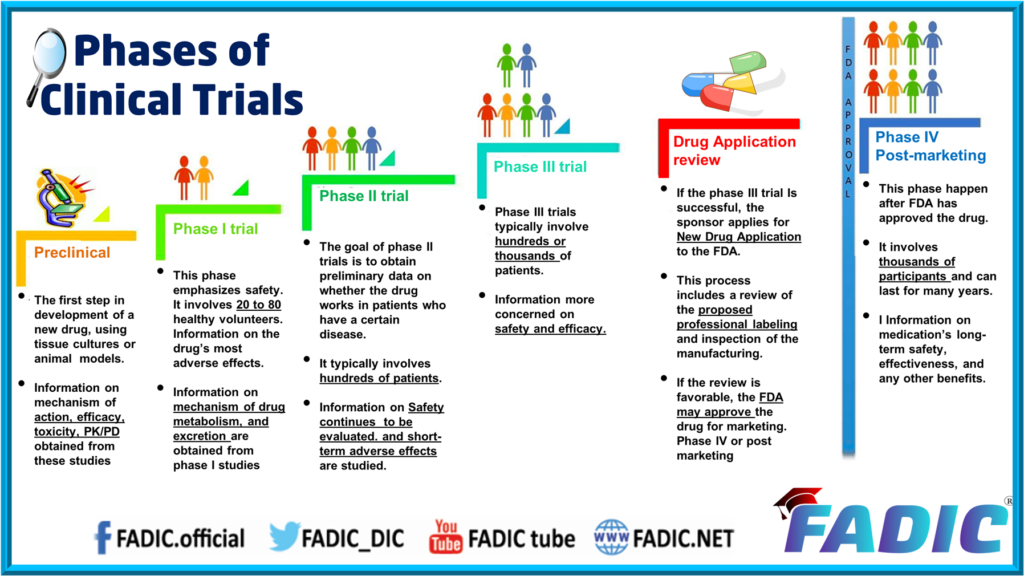
Overview of clinical trial phases
For traditional randomized designs, this depends on three primary factors that the research team must decide together – effect size, (statistical) power, and statistical significance level. Effect size refers to the minimum treatment effect that one hopes to detect in the study. Power refers to the likelihood of detecting an effect when in fact there is an effect of a priori specified size.
Clinical trials: design, endpoints and interpretation of outcomes
CDER soliciting comments regarding clinical trial innovation - FDA.gov
CDER soliciting comments regarding clinical trial innovation.
Posted: Tue, 17 Oct 2023 07:00:00 GMT [source]
Several of the challenges of clinical trial design discussed here are applicable to PLP research. Each design choice has implications for the quality and validity of your results. This course provides you and your team with essential skills to evaluate options, make good design choices, and implement them within your trial. You’ll learn to control for bias, randomize participants, mask treatments and outcomes, identify errors, develop and test hypotheses, and define appropriate outcomes. Finally, a trial without participants is no trial at all, so you’ll learn the guiding principles and develop the essential skills to ethically and conscientiously recruit, obtain consent from, and retain trial participants.
Preliminary data may come from models, observational studies or the endpoints of related studies. Some preliminary data should be available to support most of the trial’s design; not only to support the hypothesis but also to support trial feasibility. It is essential to learn as much as possible about the target disease’s epidemiology (e.g. prevalence), and about recruitment and adherence in similar trials.
Clinical trial design is a key aspect of the successful conduct of clinical trials. To address this need, in March 2020 we began planning a phase 2 adaptive platform trial, I-SPY COVID (Fig. 1). Given the large number of potential therapeutic approaches being proposed, the study was designed to rapidly evaluate and prioritize promising agents for further phase 3 testing.
On the other hand, retrospective cohort studies identify a population with and without the risk factor/exposure based on past records and then assess if they had developed the disease/outcome at the time of study. Thus, the study design for prospective and retrospective cohort studies are similar as we are comparing populations with and without exposure/risk factor to development of outcome/disease. If risks are very low and the study is easy (and not too expensive) to conduct, a pilot trial can help quantify and better evaluate the benefits, helping optimize them for future trial designs. For instance, a study evaluating the analgesic effects of gabapentin enrolled 24 PLP patients (17). However, pain level differences in the active group (even if over half the participants had a meaningful decrease in pain) did not reach significance compared to placebo. Related to randomization, allocation concealment (which should always be implemented), also protects against selection bias, e.g. such that investigators cannot encourage only patients they think will benefit to enroll in the trial.
3 Randomization and stratification
The investigator needs to be cautious and conservative in his/her choices given the risk of losing time and resources in an invalid or valid negative trial. Progressive trial phases require more preliminary data to increase the potential benefits and reduce the risks of the trial. Poorly designed trials often lead to inconclusive or negative results, usually at significant human and financial cost. Clinical trials can only be ethically justified when the potential benefits of a new intervention (whether it is a drug, device, surgery, etc.) for the target population outweighs its risks (4,5). Benefits are “the positive results of a given treatment for an individual or a population (i.e. efficacy, convenience, or even quality of life)”, while risks are “the unfavorable negative results (adverse outcomes) of a given treatment for an individual or a population” (6). The expected risk/benefit ratio will depend on the trial phase and supporting data, as we discuss below.
Seasonal studies
For example, toxic epidermal necrolysis, where clinical outcomes in cyclosporine treated patients can be compared with historical controls treated in the same center with IVIg in the past. In the treatment of some diseases, it may take a very long time to observe the definitive endpoint (e.g., death). A surrogate endpoint is a measure that is predictive of the clinical event but takes a shorter time to observe. The definitive endpoint often measures clinical benefit whereas the surrogate endpoint tracks the progress or extent of disease. Surrogate endpoints could also be used when the clinical end-point is too expensive or difficult to measure, or not ethical to measure. Other examples of subjective endpoints include depression, anxiety, or sleep quality.
It is the role of the investigator to optimize trial design and thereby maximize the potential benefits of the scientific investigation. The ethical limitations of a placebo control are partially overcome by a cross over design in which each patient receives both interventions but in a different order. Each person serves as his/her own control results in balancing the covariates in treatment and control arms. In this design, some participants start with drug A and then switch to drug B (AB sequence) in one trial arm, while subjects in other trial arm start with drug B and then switch to drug A (BA sequence). Adequate washout period must be given before crossover to eliminate the effects of the initially administered intervention. Outcomes are then compared within the same subject (effect of A vs. effect of B).
The researcher needs to perform a benefit/risk assessment when choosing a study design. Trial results can never be guaranteed but it may be reasonable to expect benefit based on previous studies; this benefit may occur at the individual or group level, with subsets of patients improving dramatically and others not at all. A critical element of a non-inferiority design is the non-inferiority margin the design will exclude. There are no specific rules on what threshold would warrant a conclusion of “non-inferiority,” though some regulatory agencies have provided guidance in some situations, and margins vary widely across endpoints, patient populations, and trials [39,40,41,42,43,44].














